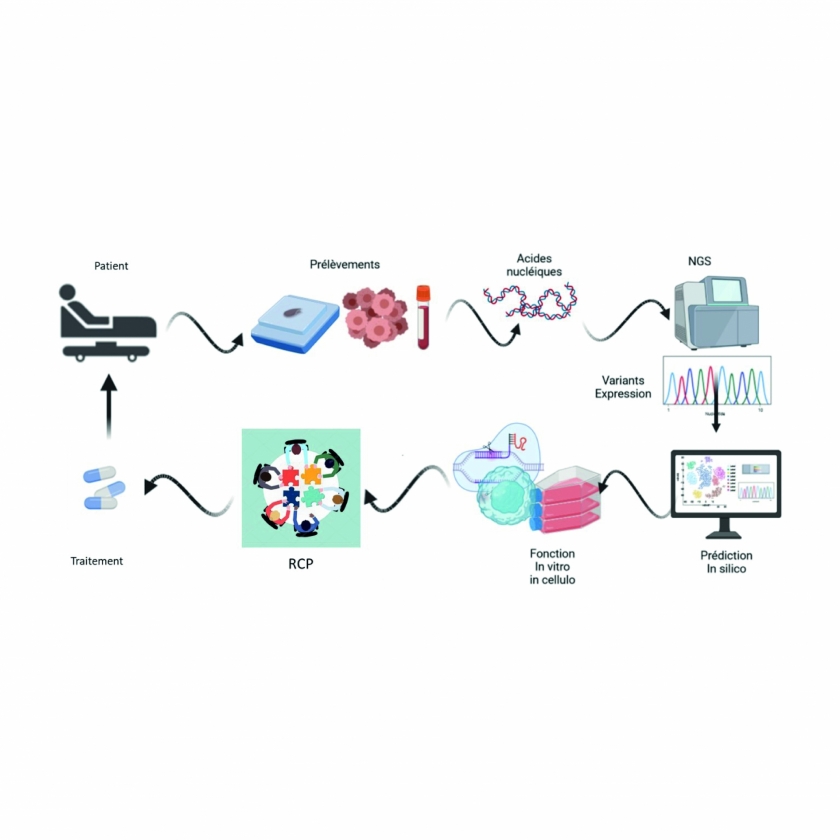
Caractérisation de l’instabilité génomique
L’unité met en place toutes méthodes diagnostiques de caractérisation moléculaire des variants génomiques en fonction des biomarqueurs indispensables pour l’éligibilités des patients aux différents traitements ciblés disponibles. On peut ainsi citer entre autres, la mise en place de séquençage NGS multi-cibles pour les inhibiteurs de tyrosine kinase dans les cancers du poumon (recommandation INCa), d’un panel de gènes du système de recombinaison homologue pour les cancers de l’ovaire et du sein pour l’administration des Inhibiteurs de PARP (projets Great et Dolaf). La mise en place de séquençage d’ARN tumoral pour la détection des gènes de fusion (Projet INCa). Dans le cadre des prescriptions d’immunothérapie l’unité détermine l’instabilité génétique dans différentes localisations. En outre, l’unité réalise les séquençages d’exomes et de transcriptomes pour le projet multi-omique Epicure Sein (https://projet-epicure.fr/) et prochainement Epicure poumon.